Le déficit en hormone de croissance chez l`enfant

THÉMATIQUE À TAPERBIOLOGIE HORMONALE
REVUE FRANCOPHONE DES LABORATOIRES - AVRIL 2009 - N°411 // 63
Le décit en hormone de croissance
chez l’enfant : formes cliniques
et biologiques
a
Service d’endocrinologie
c
Laboratoire central
Centre hospitalo-universitaire Bab El Oued
02, bd Said Touati Bal El Oued 16000
Alger – Algérie
b
Service d’endocrinologie
Hôpital Bologhine
Bains Romains 16600
Alger – Algérie
* Correspondance
haddam25@yahoo.fr
article reçu le 29 octobre 2008, accepté le 17 février 2009.
© 2009 – Elsevier Masson SAS – Tous droits réservés.
RÉSUMÉ
Le décit en GH est une étiologie rare de retard staturo-pondéral (RSP)
(13,27 %). Son diagnostic est tardif (8,48 ± 4,3 ans chez le garçon et
6,9 ± 3,8 ans chez la lle). Le retard statural est sévère (-4,7 ± 1,8 DS chez
les garçons ; - 4,9 ± 2,3 DS chez la lle). Le bilan hormonal a conrmé le
diagnostic positif par la non réponse aux tests de stimulation de la GH
(tests au glucagon-propranolol et à l’insuline) et le dosage de l’IGF1 signi-
cativement abaissés. La recherche d’autres atteintes hypophysaires a
révélé une insufsance antéhypophysaire multiple dans 49 %.
Retard staturo-pondéral – déficit en hormone de croissance –
insuffisance antéhypophysaire – GH.
L dé i GH é i l i d d dé l (RSP)
SUMMARY
Growth hormone deficiency (GHD) in child :
clinical and biological aspects.
Statural delayed is very frequent in paediatric
and endocrinology consultation. Growth hor-
mone deciency (GHD) is rarely (13,27%). The
diagnosis is delayed (8,48 ± 4,3 years for boys
and 6,9 ± 3,8 years for girls). The statural de-
ciency is severly (-4,7 ± 1,8 SD for boys and
-4,9 ± 2,3 SD for girls).
No response to stimulated tests (glucagon-
propranolol and insulinemia) and low IGF1 conr-
med the GHD.
Multiple hormonal deciencies are associated
in 49%.
Statural delayed – growth hormone deficiency –
pituitary insuffisency – GH.
Nora Soumeya Fedala
a, Ali El Mahdi Haddam
b, Akila Zenati
c, Farida Chentli
a
1. Introduction – Généralités
Le retard statural est un motif fréquent de consultation [1, 2].
Ses étiologies sont nombreuses et leur fréquence respective
diffère quelque peu selon que l’on soit en endocrinologie
ou en pédiatrie. Bien que la pathologie endocrinienne ne
soit en cause que dans un peu moins de 10 % des cas [3,
4], sa reconnaissance est primordiale car elle conduit à un
traitement spécique qui améliore le pronostic statural.
Le décit en hormone de croissance (GHD) est une cause
rare de RSP [1, 4, 5]. Sa prévalence exacte est méconnue
dans les pays émergents comme l’Algérie. En Europe et
aux États-Unis, elle est diversement estimée et varie entre
1/4 000 et 1/10 000 [4, 6]. Cette variabilité s’explique par le
polymorphisme clinique, les limites des tests de stimulation
de l’hormone de croissance et les problèmes d’interpréta-
tion de la valeur seuil diagnostique, puisque ce n’est qu’en
1998, soit près d’un siècle après les premières descriptions
du GHD que le consensus sur les valeurs référentielles a
été établi [7].
Dans notre pays, de nombreux écueils entravent la démar-
che diagnostique et thérapeutique. Pourtant, l’absence
de diagnostic précoce et de traitement adéquat ont des
conséquences néfastes : la petite taille dénitive, avec le
retentissement psychologique qui en découle. De plus, il
faut également souligner que les patients décitaires en
GH sont connus pour avoir une espérance de vie réduite à
l’âge adulte en raison d’une augmentation de la fréquence
des cardiopathies ischémiques [8, 9] et des risques accrus
de fractures par ostéoporose [10, 11] autant des problèmes
de santé publique qui appellent à un diagnostic aussi pré-
coce que possible et à une prise en charge thérapeutique
efcace an de réduire les coûts directs et indirects liés aux
conséquences du décit en GH.
Les objectifs de cette étude ont été d’évaluer la fréquence
des GHD parmi les étiologies des RSP en consultation
d’endocrinologie et d’en analyser les différents tableaux
clinico-biologiques.
2. Population et méthodologie
2.1. Population
693 enfants (470 garçons, 223 lles) et adolescents impu-
bères d’âge inférieur ou égale à 17 ans ont été adressés

64 // REVUE FRANCOPHONE DES LABORATOIRES - AVRIL 2009 - N°411
un faisceau d’arguments cliniques et paracliniques [7] :
1. les données anamnéstiques et cliniques caractéristiques
d’insufsance somatotrope ;
2. les bilans radiologiques (AO) ;
3. les bilans biologiques non hormonaux d’orientation héma-
tologique, glycémique et lipidique notamment les différentes
fractions du cholestérol. Ces derniers ont été refaits après
substitution hormonale thyroïdienne et/ ou surrénalienne si
ces décits hormonaux existaient ;
4. le taux bas de l’IGF1 et la réponse négative aux deux
tests pharmacologiques de stimulation de la GH précé-
demment cités.
Une analyse minutieuse des données cliniques et paracli-
niques a été effectuée précisant :
- le déroulement de la période néonatale, les circonstances
du diagnostic, la sévérité du tableau clinique de décit en
appréciant le degré du retard staturo-pondéral et en calculant
la vitesse de croissance annuelle. Celle-ci étant impossible
à réaliser chez tous les patients, il a fallu tenir compte de
la vitesse de croissance au cours du suivi avant la mise en
route du traitement substitutif hormonal ;
- les signes caractéristiques du décit en GH tels le visage
poupin, l’adiposité abdominale, la chevelure ne, l’acromi-
crie, le micropénis avec ou sans cryptorchidie et l’existence
de malformations congénitales.
D’autres atteintes hypophysaires ont été recherchées :
les signes d’hypothyroïdie (bradycardie, constipation,
asthénie…), les signes d’insufsance cortico-surrénlienne
(asthénie, hypotension artérielle…), les signes de défi-
cit gonadiques telles la présence d’un micropénis, une
cryptorchidie chez les garçons pré-pubères et l’absen-
ce de caractères sexuels secondaires évalués à partir
de la classication de Tanner [13] chez les garçons dont
l’âge osseux est supérieur à 14 ans et les lles dont l’âge
osseux est supérieur à 12 ans. L’existence d’atteinte post-
hypophysaire par l’existence d’un syndrome polyuro-
polydipsique apprécié en fonction des chiffres des boissons
et diurèses théoriques pour l’âge [14].
En plus de l’exploration de l’axe GH-IGF1, la groupe GHD
a bénécié d’une étude de :
- la fonction corticotrope par les dosages statiques de cor-
tisol sanguin et d’ACTH. La réserve corticotrope est étudiée
grâce au test à l’hypoglycémie insulinique. L’atteinte corti-
cotrope est attestée par un taux bas de cortisol plasmatique
ou une réponse basse au test à l’insuline associée à une
ACTH non élevée ;
- la fonction thyroïdienne grâce aux dosages de la
thyroxinémie (FT4) et de la TSHus. À défaut de TRH non
disponible en Algérie, la réserve thyréotrope n’a pas été
appréciée. Le décit thyréotrope est attesté par un taux
bas de FT4 corrélé à une TSHus non élevée ;
- la fonction prolactinique par le dosage statique de la
prolactine. La réserve prolactinique n’a pas été étudiée
faute de TRH ;
- l’atteinte gonadique est recherchée chez les patients ayant
un âge osseux de type pubertaire grâce aux dosages bas
des stéroïdes sexuels et de leurs gonadotrophines hypo-
physaires (FSH, LH). La réserve gonadotrope n’a pu être
étudiée en raison de la non disponibilité du réactif LHRH
en Algérie. Les résultats ont été comparés aux valeurs de
référence de Roger [15] ;
en consultation d’endocrinologie entre 2001 et 2006 pour
retard statural déni par une taille inférieure ou égale à
– 2 DS par rapport à la moyenne des tailles sur les courbes
de Sempé et Pedron [12]. Tous ces patients ont bénécié
d’un interrogatoire minutieux, d’un examen clinique le plus
complet possible et d’un ensemble d’explorations paraclini-
ques (radiographie du poignet et de la main gauche de face,
formule numération sanguine, bilan rénal, glycémie à jeun,
bilan hépatique, bilan lipidique, ionogramme sanguin, bilan
phosphocalcique, vitesse de sédimentation, protides totaux,
copro-parasitologie des selles, dosage des anticorps anti-
réticulines et anti-endomysium, biopsie jéjunale, thyroxine
libre (FT4) et thyrostimuline (TSHus). Au terme de cette
première exploration, nous avons pu ainsi dégager deux
groupes de sujets ; ceux ayant des éléments en faveur d’une
étiologie non endocrinienne ou endocrinienne autre que le
décit en GH et ceux ayant une insufsance somatotrope
probable. Ces derniers ont bénécié d’un test au glucagon/
propranolol. Ce test a été systématiquement sensibilisé par
les stéroïdes sexuels chez les enfants en âge péri-pubertaire
(âge chronologique ≥ 08 ans) : 1 cp /j de valerate d’œstradiol
(2 mg) pendant 3 jours avant la pratique du test chez les
lles et 100 mg d’enanthate de testostérone en IM 3 jours
avant le test chez les garçons.
Deux groupes de sujets comparables pour l’âge et le sexe
ont pu être sélectionnés :
- le groupe G1 composé de 101 enfants (51 garçons et
50 lles) présentant un retard constitutionnel et/ou familial
et servant de groupe témoin pour l’évaluation hormonale
des patients de l’étude ;
- le groupe GHD très suspect de décit somatotrope consti-
tué de 107 patients.
Ces deux groupes G1 et GHD ont bénécié d’un 2e test de
stimulation pharmacologique de la GH : le test à l’insuline
pour répondre aux critères diagnostiques de GHD. Ce test
a aussi été sensibilisé par les stéroïdes sexuels chez les
enfants en péri-puberté. Les deux tests utilisés par l’éva-
luation de la fonction somatotrope sont les seuls utilisés
en Algérie.
Ainsi, le groupe G1 a servi de groupe témoin en comparai-
son avec le groupe GHD pour l’évaluation hormonale de la
fonction anté-hypophysaire.
Le choix des RSP constitutionnels et/ou familiaux comme
sujets témoins nous a paru judicieux étant donné que les
tests pharmacologiques de stimulation de la GH ne sont
pas, sur le plan éthique, envisageables chez l’enfant sain
et que pour attester d’une fonction somatotrope normale,
nous avons recours à ces testes dynamiques.
Par ailleurs, comme il a été proposé d’étudier l’IGF1 chez
les enfants GHD, il s’est avéré indispensable d’ajouter un
deuxième groupe d’enfants témoins sains de taille normale
pour comparer les dosages de l’IGF1 avec ceux du groupe
GHD. Ce groupe appelé G2 est constitué de 266 enfants
sains indemnes de toute affection pouvant retentir sur l’axe
GH-IGF1. Ils ont été regroupés par tranche d’âge et selon
leur stade pubertaire en se basant sur la classication de
Tanner [13].
2.2. Méthodologie
Il s’agit d’une étude analytique, comparative prospective et
longitudinale des 107 GHD dont le diagnostic a été établi sur
REVUE FRANCOPHONE DES LABORATOIRES - AVRIL 2009 - N°411 // 65
BIOLOGIE HORMONALE
lles (p < 0,02). Faute de carnets de santé, correctement
tenus, les paramètres auxologiques à la naissance n’ont
pas été documentés en totalité (2/3 pour le poids, 1/3 pour
la taille). La taille moyenne et le poids moyen de patients
GHD nés à terme sont normaux pour l’âge gestationnel
sans différence signicative (tableau II).
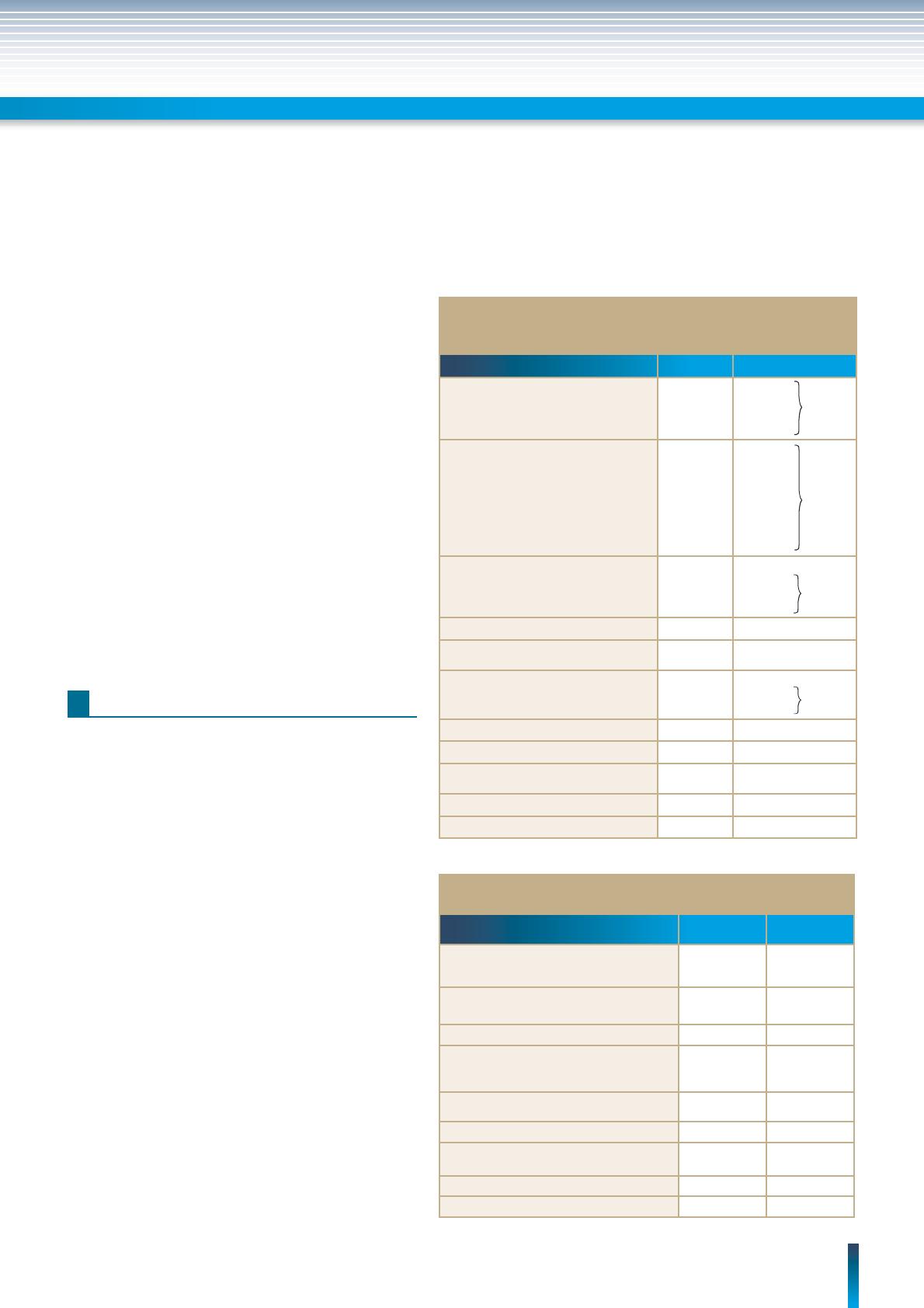
Tableau I – Répartition de l’ensemble des RSP (n = 693)
en fonction des étiologies observées au niveau
d’une consultation d’endocrinologie.
Étiologies Nombre %
- R constitutionnel
+ R pubertaire
- R familial
+ R pubertaire
194
22
157
5
28
3, 17
22, 65, 54
0,72
Affections digestives
Affections hématologiques
Affections neurologiques
Affections osseuses
Affections respiratoires
Affections rénales
Cardiopathie
Hépatique
99
16
7
7
4
4
3
1
14,28
2,30
1,01
1,01
0,57
0,57
0,43
0,14
Endocrines
GHD
Hypothyroïdie primaire
Hypoparathyroïdie
92
22
02
13,27
3,17
0,28
RCIU 21 3,03
Affections chromosomiques
(syndrome de Turner + trisomie 18) 14 2,02
Affections métaboliques
Diabète sucré
Maladie de Wilson
12
1
1,73
0,14
Syndrome polymalformatif 8 1,15
Corticothérapie 1 0,14
Autres étiologies (mucoviscidose,
mucopolysaccharidose….) 00
Malnutrition 1 0,14
Total 693 100
54 %
20,34 %
16,73 %
1,87 %
Tableau II – Caractéristiques cliniques
des patients GHD à la naissance dans les deux sexes.
Patients Garçons
(n = 62 )
Filles
(n = 45)
Âge gestationnel
À terme
Prématurité
59
3
42
3
Dystocie néonatale
Siège
14 (22,4)
7 (11,3)
3 (6,7)
1 (2,2)
Souffrance néonatale 12 (19,3) 2 (4,4)
Taille moyenne à la naissance à terme
et documentée (n = 30)
X cm ± SD (extrêmes )
50,3 ± 2,1
(48 – 56)
49,7 ± 1,1
(48 – 52)
Poids moyen à la naissance (extrêmes) 3,5 ± 0,7
(1,5 – 5)
3,2 ± 0,8
(2 – 5)
Anomalies de la ligne médiane 30 (48,4) 10 (22,2)
Micropénis
Micropénis + cryptorchidie uni ou bilatérale
21 (33,3)
15 (24,2)
Ictère 2 2
Hypoglycémie 3 (4,8) 1 (2,2)
- l’atteinte post-hypophysaire est mise en évidence lorsqu’il
existe un syndrome polyuro-polydipsique [14] associé à une
densité urinaire inférieure à 1005. L’absence de son élévation
lors du test de restriction hydrique et après administration
d’analogue de l’hormone antidiurétique afrme le diabète
insipide. Une fois le diagnostic de GHD établi, une hormo-
nothérapie substitutive par la GHr a été indiquée à raison
de 0,7 u/kg/semaine en sous cutané 7/7j. Ce traitement a
été associé en cas de décits hypophysaires multiples à de
l’hydrocortisone = 5 à 10 mg/m/j et/ou de la levothyroxine :
2,5 ng/kg/j par voie orale.
2.3. Étude statistique
Les résultats ont été analysés sur ordinateur comportant
un logiciel Epi Info version 6.4 après enregistrement des
données. Les méthodes utilisées sont :
- le calcul des moyennes arithmétiques (X ± SD). La
comparaison entre deux moyennes est basée sur le test
de student et l’écart réduit ;
- le calcul des pourcentages arithmétiques (p %). La
comparaison entre 2 pourcentages est basée sur le test
du chi 2 et l’écart réduit. Le taux de significativité est
p < 0,05.
2.4. Techniques et normes
Les dosages hormonaux ont été réalisés par deux
techniques radio-immunologiques (RIA) et immunora-
diométriques (IRMA) sauf pour l’ACTH qui est dosée par
chimiluminescence.
3. Résultats
3.1. Fréquence relative des GHD
pour les autres étiologies en consultation
d’endocrinologie
Près de 3/4 des RSP (83,2 %) consultant en endocrino-
logie ne relèvent pas d’une étiologie endocrinienne. La
moitié (54,54 %) d’entre eux sont des RSP constitution-
nels et familiaux associés ou non à un retard pubertaire.
20 % des RSP sont liés aux affections viscérales domi-
nées par la malabsorption digestive dans 3/4 des cas.
La cause endocrinienne est présente dans 16,7 % des
cas. Le décit en GH est présent 8 fois sur 10 (13,27 %)
dans une proportion relativement égale avec la pathologie
digestive (tableau I).
3.2. Caractéristiques cliniques des patients
GHD à la naissance dans les deux sexes
Une prédominance masculine classique est notée (sex
ratio G/F : 1,38). La majorité des patients (94,4 %) sont
nés à terme. Seulement 5,6 % sont nés prématurément
(VS 11,2 %, population algérienne DNS) Une dystocie
néonatale n’a été retrouvée que dans 15,9 % des cas
(vs 15,34 % population algérienne (DNS). Dans près de
la moitié des cas, la dystocie est liée à un accouchement
par le siège (n = 8/17 ; 47,1 %). La souffrance néonatale
n’a été constatée que dans 13,1 % des cas (vs 7,8 %
DNS) (tableau II).
Les événements périnataux (dystocie, souffrance) sont
retrouvés plus fréquemment chez les garçons que chez les

66 // REVUE FRANCOPHONE DES LABORATOIRES - AVRIL 2009 - N°411
3.3. Caractéristiques cliniques
des GHD à la première consultation
dans les deux sexes
L’âge moyen au moment du diagnostic de patients GHD
est tardif. Les garçons GHD consultent un peu plus tardi-
vement par rapport aux lles ; cette différence est à la
limite de la signicativité (p = 0,059). Le délai moyen entre
la constatation du retard statural et la première consulta-
tion est de 4,17 ans
±
3,29 ans. Ce délai moyen est signi-
cativement plus important chez les garçons que chez les
lles (p < 0,005) (tableau III).
Le retard statural est sévère chez les patients GHD qu’il
soit exprimé en déviations standards ou en score dévia-
tions standards et sans différence signicative entre les
deux sexes (DNS). Ce retard est aussi sévère lorsqu’il
est analysé par rapport à la taille cible parentale comme
pour la taille, il existe un retard pondéral important. Ce
retard est cependant, moins sévère que le retard statural
(p < 10-6). Un nanisme est noté dans plus de 2/3 des cas
(78 %) sans différence signicative entre les deux sexes
(p = 0,84). Le morphotype est typique quel que soit le sexe
et caractéristique du décit en GH dans la majorité des
cas (88,8 %) (tableau II).
3.4. Résultats paracliniques
des patients GHD dans les deux sexes
à la première consultation
3.4.1. Bilan non hormonaux
Il existe un retard important de l’âge osseux chez tous les
patients GHD sans différence signicative entre les deux
sexes (DNS) (tableau IV).
Une anémie est retrouvée dans un peu moins de la moitié
des cas (41,11 %) majoritairement microcytaires (86,36 %)
(tableau IV).
La glycémie moyenne est normale sans différence signi-
cative avec la valeur moyenne occidentale. Une glycémie
normale basse < à 0,65 g/l a été retrouvée dans 11,2 %
des cas.
Les valeurs moyennes de la fraction HDL du cholestérol
et des triglycérides sont aussi normales sans différence
signicative avec les valeurs normales théoriques. Seule la
valeur moyenne du cholestérol total est signicativement
abaissée par rapport aux valeurs normales théoriques
occidentales (tableau V).
3.4.2. Bilans hormonaux (tableau VI)
Le pic de GH moyen des patients GHD est bas aussi
bien pour le test au glucagon/propranolol que pour le test
à l’insuline avec une différence hautement signicative
entre les pics moyens des GHD et des témoins (p < 10-6).
Dans le groupe G1, la réponse de la GH au test combiné
glucagon/propranolol est plus ample et plus importante
que lors du test simple à l’insuline.
Avant 4 ans, il existe un chevauchement des résultats entre
les IGF1 des enfants GHD (n = 5) et les IGF1 des enfants
témoins G2 du même âge. Après 4 ans, les taux d’IGF1
sont signicativement abaissés pour les différentes tranches
d’âge des sujets GHD comparativement au groupe G2 et
ce quelque soit le stade pubertaire (tableau VII).
Tableau III – Caractéristiques cliniques des GHD
à la première consultation dans les deux sexes.
Garçons
(n = 62)
Filles
(n = 45)
Âge moyen
(X ans ± SD)
(extrêmes)
8,5 ± 4,3
(0,16 – 18)
6,9 ± 3,8
(0,83 – 16,58)
Délai moyen entre la constatation du RS
et la 1re consultation
X années ± SD
(extrêmes)
4,9 ± 3,5
(18 J – 14 ans)
3,1 ± 2,7
(18 J – 12 ans)
Taille moyenne
X DS ± SD
(extrêmes)
X SDS ± SD
(extrêmes)
- 4,7 ± 1,8
(- 12,0)
- 4,7 ± 1,6
(- 9,4 ; 0,8)
- 4,9 ± 2,3
(- 11,9 – - 2,8)
- 4,9 ± 2,3
(- 11,9 – 2,9)
Taille cible parentale
XDS ± SD
(extrêmes)
-3,7 ± 2,3
(- 10,5 ; - 0,9)
-4,6 ± 2,6
(-13 – - 1,5)
Poids moyen
X DS ± SD
(extrêmes)
X SDS ± SD
(extrêmes)
- 2,6 ± 1,1
(- 5 – 0)
- 2,8 ± 1,2
(- 4,8 – - 3,9)
- 2,9 ± 0,8
(- 5 – - 0,5)
- 2,9 ± 1,1
(- 5,9 – - 0,3)
Morphotype
Typique
Atypique
55 (88,7)
7 (11,3)
40 (88,9)
5 (11,1)
Tableau IV – Résultats paracliniques osseux et hématologiques
des patients GHD au diagnostic dans les deux sexes.
Âge osseux Garçons Filles Résultats
Âge osseux moyen
X années ± SD
(extrêmes)
4 ± 2,8
(0,25 – 11)
3,4 ± 2,3
(0,5 – 8)
3,8 ± 2,6
(0,25 – 11)
Retard de l’Âge osseux (AO / AC)
X mois ± SD
(extrêmes)
0,5 ± 0,2
(0,14 – 1,5)
0,5 ± 0,2
(0,13 – 0,9)
0,5 ± 0,2
(0,13 – 1,5)
FNS
Anémie
- Microcytaire
- Normochrome
- Hypochrome
- Normocytaire
- Hypochrome
- Normochrome
23
28
15
10
4
3
1
21
10
4
9
2
2
-
44 (41,1)
38 (35,5)
19 (17,8)
19 (17,8)
6 (5,6)
5 (4,7)
1 (0,9)
Tableau V – Résultats métaboliques glucido-lipidiques
des patients GHD au diagnostic dans les deux sexes,
comparaison avec une référence occidentale [17].
Paramètres Valeurs
sériques
Valeurs
référen-
ces occi-
dentales
P
Glycémie
Xg ± SD
(extrêmes)
0,8 ± 0,1
(0,8 – 0,8) 0,7 – 1 DNS
Triglycérides
Xg ± SD
(extrêmes)
0,8 ± 0,3
(0,7 – 0,9) 0,5 – 1,5 DNS
Cholestérol total
Xg ± SD
(extrêmes)
1,7 ± 0,3
(1,6 – 1,7) 1,6 – 2,4 P < 0,05
HDL cholestérol
Xg ± SD
(extrêmes)
0,5 ± 0,2
(0,5 – 0,5)
≥ 0,5 M
≥ 0,6 F DNS
LDL cholestérol
Xg ± SD
(extrêmes)
1,0 ± 0,3
(1,0 – 1,1) < 1,6 DNS

REVUE FRANCOPHONE DES LABORATOIRES - AVRIL 2009 - N°411 // 67
BIOLOGIE HORMONALE
Par ailleurs, les valeurs d’IGF1 retrouvées dans le groupe
témoin constitué d’enfants sains sont inférieurs signica-
tivement à celles des enfants sains occidentaux de taille
normale à l’exception de la tranche d’âge 8-10 ans ou les
valeurs de l’IGF1 ne sont pas différentes signicativement
(tableau VIII).
Sur les 107 patients GHD, 62,61 % (n = 67) étaient en
eucortisolisme ; 20,56 % (n = 22) avaient une insufsance
corticotrope de réserve et 16,82 % (n = 18) présentaient
un hypocorticolisme basal malgré l’altération de l’axe
corticotrope dans 2/3 des cas ; l’insufsance corticotrope
était asymptomatique dans la majorité des cas (90 %) chez
les patients eucortisoliques. La valeur moyenne basale
de l’ACTH est normale dans le groupe GHD et signi-
cativement abaissée par rapport à celle du groupe G1
(p < 0,008). En revanche, les valeurs moyennes du corti-
sol plasmatique basal du groupe GHD et du groupe G1
ne sont pas différentes sur le plan statistique, il en est de
même pour ∆ cortisol (tableau VI). Les valeurs moyennes
de l’ACTH et du cortisol plasmatique de base sont nor-
males sans différence signicative avec celles du groupe
témoin G1. En revanche, il existe une différence haute-
ment signicative entre le ∆ cortisol des patients GHD et
celui du groupe G1 (p < 0,008) (tableau VI). Les valeurs
moyennes de l’ACTH plasmatique et du cortisol moyen
basal des patients GHD ayant une insufsance corticotrope
patente sont signicativement abaissées par rapport au
groupe G1 (tableau VI).
Un tiers des patients GHD (31,70 %) avaient une hypo-
thyroïdie patente. Les autres patients (48,6 %) avaient
une fonction thyroïdienne normale à l’état statique. 7 fois
sur 10 les patients ayant une hypothyroïdie patente étaient
pauci-symptomatique et de diagnostic systématique au
moment de l’évaluation.
Les valeurs moyennes de la TSHus du groupe GHD hypo-
thyroïdiens et euthyroïdiens sont normales sans différence
signicative avec le groupe G1 (tableau VI). Il en est de
même pour la FT4 moyenne des patients euthyroïdiens.
En revanche, la FT4 moyenne des patients euthyroïdiens
est franchement abaissée avec une différence hautement
signicativement par rapport au groupe G1 (p < 0,0004).
Concernant l’axe lactotrope, la valeur moyenne de prolac-
tine basale des patients GHD est normale, il faut remarquer
cependant que le taux moyen de prolactine est statistique-
ment plus élevé que celui de G1 (tableau VI).
Les valeurs moyennes des gonadotrophines hypophysaires
et des stéroïdes sexuels (tableaux IX et X).
Le regroupement des décits hypophysaires au diagnos-
tic révèle que le décit en GH est isolé dans la moitié des
cas n = 54/107 (50 %) et associé à d’autres décits dans
l’autre. Moitié n = 53/107 (49 %). Le décit triple GH, TSH,
ACTH est prédominant parmi les décits hypophysaires
combinés (37,73 %). Les décits doubles GH + TSH et
GH = ACTH sont en proportions égales sans différence
signicative.
4. Discussion
Le GHD constitue un motif rare de consultation en endo-
crinologie. 107 patients colligés en 6 ans dont 15 dia-
gnostiqués avant l’étude et perdus de vue. Concernant les
Tableau VI – Résultats hormonaux,
comparaison avec les témoins G1.
Paramètres GHD (n = 107) G1 (n = 101) P
GH moyenne après glucagon
X mui/l ± SD
(extrêmes)
2,8 ± 3,2
(0,1 – 14)
47,6 ± 22,9
(19,0 – 10,1) P < 10-6
GH moyenne après insuline
X mui/l ± SD
(extrêmes)
1,7 ± 2,3
(0,1 – 9,9)
32,1 ± 23,3
(16,4 – 192,5) P < 10-6
ACTH moyenne
- Eucortisoloms (n = 67)
X mg/ml ± SD
(extrêmes)
- Hypocortisoliques (n = 22)
de réserve
Xpg/ml ± SD
(extrêmes)
-Hypocortisoliques (n=18)
Xpg/ml ± SD
(extrêmes)
16,9 ± 7,9
(6,6 – 49,3)
19,7 ± 8,0
(10 – 46,7)
15,8 ± 6,0
(10 – 28,2)
21,3 ± 9,1
(10 – 60)
21,3 ± 9,1
(10 – 60)
21,3 ± 9,1
(10 – 60)
P <0,008
DNS
P < 0,015
Cortisol sanguin moyen
- Eucortisolique (n = 67)
X nmol/l ± SD
(extrêmes)
- Hypocortisolique
de réserve (n = 22)
X nmol/l ± SD
(extrêmes)
- Hypocortisolique (n = 18)
Xnmol/l ± SD
(extrêmes)
402,01 ± 172,9
(154 – 777,5)
256,1 ± 63,1
(155,1 – 364,9)
94,1 ± 47,7
(5,9 – 144,7)
377,6 ± 159,6
(119,7 – 684,2)
377,6± 159,6
(119,7 – 684,2)
377,6 ± 159,6
(119,7 – 684,2)
DNS
DNS
P < 0,01
∆1 cortisol après insuline
- Eucortisolique (n = 67)
X nmol/l ± SD
(extrêmes)
- Hypocortisoliques
de réserve
(n = 22)
X nmol/± SD
(extrêmes)
359,7 ± 266,7
(58,9 – 1886)
140,1 ± 94,7
(155,1 – 364,9)
382,6 ± 190,8
(118,8 – 1251,8)
382,6 ± 190,8
(118,7 – 1251,8)
DNS
P < 0,008
TSH moyenne hypothyroï-
diens (n = 55)
X uU/ml ± SD
(extrêmes)
Euthyroïdiens (n = 52)
X uU/ml ± SD
(extrêmes)
FT4 moyenne
Hypothyroïdiens (n = 55)
X pmol/ml ± SD
(extrêmes)
Euthyroïdiens (n = 73)
Xpmol/ml ± SD
(extrêmes)
2,5 ± 1,4
(0,1 ± 5,9)
2,3 ± 1,1
(0,2 – 6,2)
4,7 ± 2,4
(0,9 – 7,3)
13,9 ± 3,3
(8 – 22,4)
2,3 ± 1
(0,3 – 4,6)
2,3 ± 1
(0,3 – 4,6)
14,3 ± 2,9
(10,6 – 23,8)
14,3 ± 2,9
(10,6 – 22,8)
DNS
DNS
P < 0,004
DNS
Prolactine moyenne
X mul/ml ± SD
(extrêmes)
256 ± 262,8
(0,3 – 1937,2)
196,2 ± 95
(60 – 500) P < 0,03
1 ∆ : différence entre pic maximum de cortisol après insuline et la valeur basale.
retards staturaux eux-mêmes, l’étiologie endocrinienne est
rare : 16,73 % dans cette étude, résultat proche des 10 %
reconnus habituellement [2, 8, 19, 20, 21]. Ces constata-
tions incitent à penser prioritairement aux retards consti-
tutionnels et familiaux qui à eux seuls regroupent plus de
la moitié des étiologies de RSP. Les affections digestives
doivent aussi être recherchées systématiquement devant
tout RSP. En effet, à elles seules, elles sont presque aussi
fréquentes que tous les décits endocriniens réunis 14,28 %
vs 16,73 %. La malabsorption digestive est en réalité même
un peu plus fréquente que le décit en GH dans cette étude
14,28 % vs 13,27 %. Cette proportion est expliquée par
 6
7
8
6
7
8
1
/
8
100%








